What are the cutting-edge BioInformatic tools we use?
We have developed and use the M-tools suite for microbial metagenomics and iVirus for viral metagenomics.
M-tools
The M-tools suite is software designed and developed by the Australian Centre for Ecogenomics (ACE) and are used in the analysis of ecogenomic datasets. IsoGenie Co-PI Dr. Gene Tyson, Deputy Director of ACE, uses this suite of tools to analyze the data for this project. The M-tools are a suite of metagenomic analysis tools targeted toward the recovery of high-quality population genomes from metagenomic data. The M-tools suite also includes software for constructing strain-resolved community profiles, and the analysis of gene families within unassembled metagenomic data.
- GraftM: Phylogenetically informed analysis of
gene families in meta-omic data
GraftM is a tool for the analyses of gene families within sequence data. GraftM searches metagenome sequence data using either a DIAMOND database, or a hidden Markov model (HMM) search coupled to a rapid ORF caller (OrfM, see below). GraftM classifies sequences using a phylogenetic tree insertion. - SingleM: Strain-resolved
taxonomic analysis of metagenome communities
Currently, community profiles from metagenomes lack resolution. SingleM aims to provide strain-resolved taxonomic community profiles. SingleM can be used to link OTUs to population genomes to estimate level of community recovery. SingleM is currently being developed on GitHub. - GroopM: Differential coverage based binning
of population genomes
As we enter a period of genome-centric analysis of metagenomes, accurate, fast population genome recovery methods are needed. GroopM is a metagenomic binning toolset leveraging differential coverage to extract population genomes from multi-sample metagenomic datasets. Once you have your genomes, you can use our companion tool CheckM to assess the completeness and contamination of recovered genomes. For a publication about this tool – GroopM: an automated tool for the recovery of population genomes from related metagenomes. - CheckM:
Lineage specific marker set based quality control of population genomes. CheckM provides a set of tools for assessing the quality of genomes recovered from isolates, single cells, or metagenomes. It provides robust estimates of genome completeness and contamination by using collocated sets of genes that are ubiquitous and single-copy within a phylogenetic lineage. For a publication about this tool – CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. - RefineM: Analysis
and refinement of population genomes
Once recovered, population genome bins with contamination of low completeness require some degree of refinement. RefineM provides the tools for visualising and analysing quantitative and qualitative features of contigs in order to remove potential contaminants. The completeness of population genome bins can be further improved by recruiting contigs with similar coverage, GC percentage and tetranucleotide frequency. RefineM may also be used as a companion tool to CheckM to identify population genomes that should be merged together. RefineM is currently being developed on GitHub. - OrfM: Not-slow
open reading frame finder
OrfM is a rapid open reading frame predictor of metagenomic data. OrfM avoids six-frame translation of nucleotide sequences by first identifying stop codons using the Aho-Corasick algorithm. OrfM performs faster than other similar programs, allowing it to scale to growing metagenomic datasets. OrfM is currently being developed on GitHub.
iVirus
The iVirus project (ivirus.us) is a set of software tools, data repository, and metadata resources built on the NSF-funded CyVerse cyberinfrastructure (cyverse.org). Co-led by IsoGenie co-PIs Matthew Sullivan at OSU and Bonnie Hurwitz at UA, the project focuses on addressing challenges unique to viral ecology and biology, with many tools specific for viruses and metagenomics. Leveraging CyVerse, researchers can access shared data, common compute resources, and a platform for developing and using apps – packaged software tools build within a user-friendly interface. These apps cover nearly every stage of the viral analysis pipeline, including processing raw sequence data, assembly, post-assembly filtering, and sequence analysis. View the iVirus protocols here.
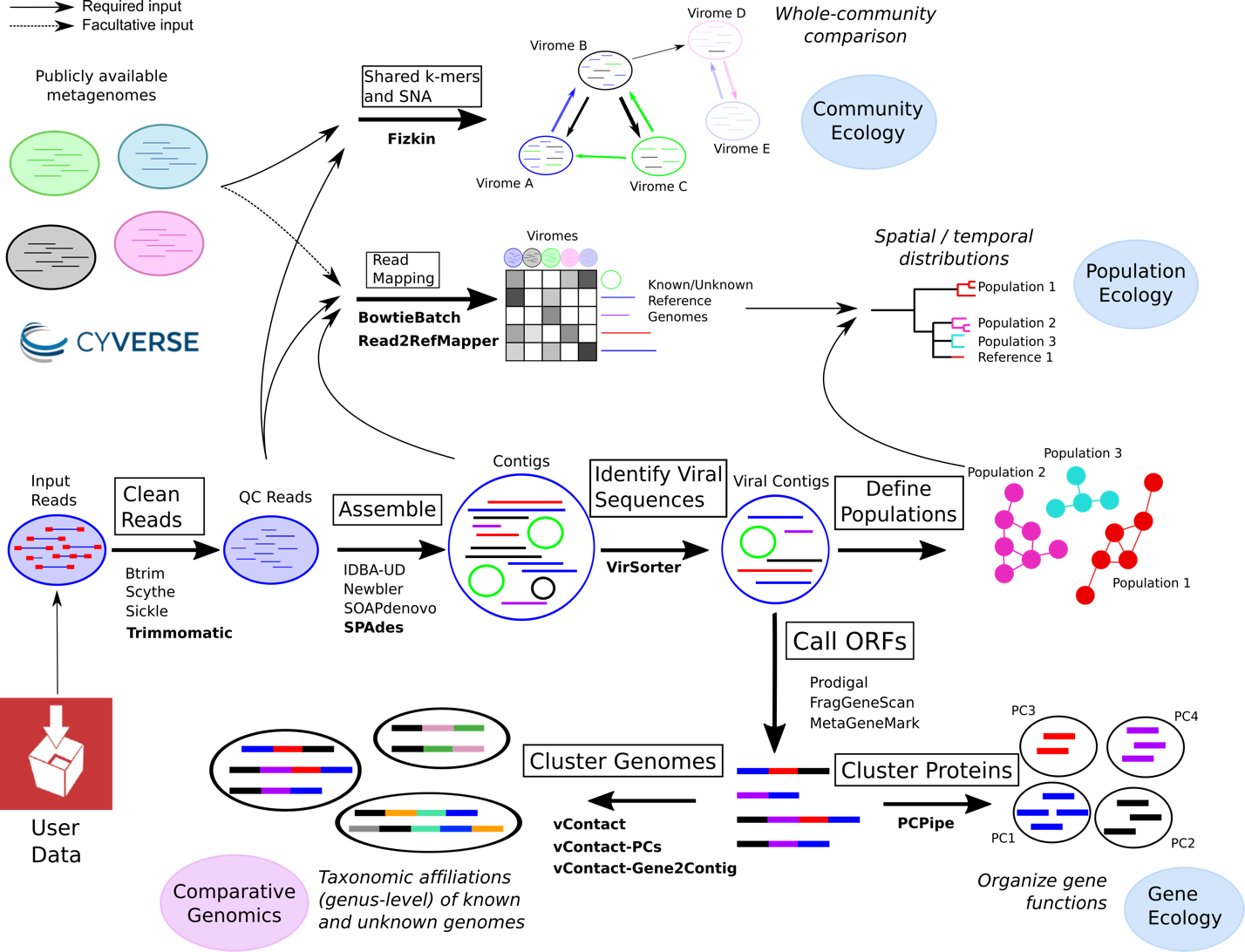
Fig 1: An overview of how a user can leverage iVirus and CyVerse apps to analyze a viral metagenomic dataset.
Viral Analysis
- PCPipe: This app compares open reading frames (ORFs) from a user-defined dataset to existing viral protein clusters (PCs) as a means to organize proteins derived from viral metagenomics into functional units that can be used as 1) a universal functional diversity metric for viruses 2) a scaffold for iterative functional annotations, and 3) input to ecological comparisons. Users provide user-generated ORFs from viral metagenomics assemblies, and PCPipe matches them against a user-supplied PC database, then self-clusters the remaining unclustered ORFs to capture the PCs unique to that dataset.
- VirSorter: This app identifies viral sequences in microbial genomes and metagenomic datasets. VirSorter can identify diverse viral sequences from microbial datasets, both integrated into the host chromosome and extrachromosomal. This tools is powerful and highly-scalable, as its first application was to nearly 15,000 publicly available archaeal and bacterial genomes, where VirSorter identified 12,498 new host-associated viruses and their genomes, increasing the publicly known viral genome reference datasets nearly 10-fold.
- vContact: This app assigns potential viral sequences to taxonomic groups using the presence of absence of shared protein clusters along the length of the sequence. This is critical as viruses lack a universal gene mark and less than 0l1% of viruses in natural environments are represented in public databases, arguing for new methods of taxonomically classifying novel viruses. Reference sequences and their taxonomic lineages can be seeded into the analysis to improve clustering and taxonomic predictions. Finally, vContact results in a network that can be further mined using its topological properties, with clusters roughly corresponding to genus-level ICTV classification. vContact can also incorporate annotations associated with contig and PCs, allowing users the ability to examine the relationship of any annotated contig/PC in context of its cluster and position in the network.
- vContact-PCs: Designed as a companion tool for vContact, this app generates protein clusters using the Markov clustering algorithm. Users provide a blastp file (which can be generated through the blastp app in CyVerse) of an all-against-all protein comparison, and vContact-PCs generates the necessary files for vContact.
- BatchBowtie: Inspired by users, this app runs Bowtie2 on any number of read files within a user-defined directory on CyVerse against a reference dataset. It can handle interleaved and non-interleaved fastq files, compressed files, and can generated SAM and/or BAM-formatted outputs.
- Read2RefMapper: This app uses BAM alignment files and generates coverage tables and relative abundance plots, useful for identifying the abundance of reads against a set of reference sequences. Users can select a variety of filtering options based on the percent of read and reference covered. If provided with the size of each metagenome, Read2RefMapper can normalize the coverage between samples.